

��ǰλ��: 늳�(li��n)�˾W(w��ng) > ǰ�� >
��������Li10GeP2S12�՚ⷀ(w��n)���Բ����F(xi��n)��(w��n)����ȫ�̑B(t��i)�늳�
�r�g:2023-04-03 08:52��Դ:����늳���ԴCamCellLab ����:CCL����
�c��:
��
��(d��o)�Z
��(d��o)�x���ڱ����o�C���w늽��|(zh��)�У�Li10GeP2S12������w늽��|(zh��)���п���Һ�B(t��i)늽�Һ�ĸ��x��늌�(d��o)�ʣ�12mS cm-1������ȫ�̑B(t��i)�늳����ЏV韵đ�(y��ng)��ǰ����Ȼ����Li10GeP2S12��¶�ڝ�՚��O��ˮ������H2S���w�������l(f��)�x��늌�(d��o)�ʵĴ�����½���
���⣬�@�N늽��|(zh��)����䇽��ٱ��������W(xu��)����(w��n)��������ڽ��|�����ϕ��a(ch��n)���x����ӻ�ό�(d��o)�w������Li10GeP2P2S12�ķֽ⣬�s��Li10GeP2S12��ȫ�̑B(t��i)�늳صĉ�����
01������B
���գ��Ї��ƌW(xu��)Ժ�������ϼ��g(sh��)�c�����о���Ҧϼ�y�n�}�Mͨ�^ʹ�Ú�������ķ�����(g��u)����һ�N����LiF�����ӵ�LiF@Li10GeP2S12�˚�늽��|(zh��)����ͨ�^�ܶȷ�����Փ(DFT)�C�����@�N���е�ˮ�����ܵĺ˚��Y(ji��)��(g��u)늽��|(zh��)������Ч������H2O���ӵ�������PS43-��ˮ�⡣
���⣬������LiF�����ӣ�LiF@Li10GeP2S12�����늌�(d��o)�ʽ�����һ����(sh��)����������������֦�������L���p����LiF@Li10GeP2S12�c�֮�g�ĸ�����(y��ng)�����R������ܶ������������3 mA cm-2������LiF@Li10GeP2S12�M�b��LiNbO3@LiCoO2/LiF@Li10GeP2S12/Liȫ�̑B(t��i)�늳���1 C�ı�����ѭ�h(hu��n)1000�κ����94.8%�����������ʡ�ԓ���°l(f��)���ڇ��H피��ڿ�Advanced Materials�ϡ��Tʿ������㑞鱾�ĵ�һ���ߡ�
02��(n��i)�ݱ���
ͨ��������ܛӲ��A��Փ���x��ȡ�������Li10GeP2S12��՚ⷀ(w��n)���Ե���Ч������Ȼ����������Ԫ�ز�����Ч��QLi10GeP2S12�����䇽��ٱ��������W(xu��)����(w��n)�Ć��}���҂������������w늽��|(zh��)�w��������������ӿ�����һ�N�܉�ͬ�r�M���՚�ͽ��淀(w��n)���Եķ�����
�ڸ��N���Ӱ����Ӳ����У�LiF���ϲ��H�����ژO��ˮ�ܽ�Ⱥ̓�(y��u)���Ļ��W(xu��)��(w��n)���ԣ����⣬������늌�(d��o)�ʡ�����늻��W(xu��)���ڣ�0-6.3V���ߵČ�䇽����ܣ�73.28 meV Å-2�������ԣ���������Li10GeP2S12���淴��(y��ng)�������֦�����L�����ڴˣ�������ͨ�^����������Li10GeP2S12�w������������LiF�����ӡ�
1. LiF@Li10GeP2S12�ĺ˚��Y(ji��)��(g��u)����

�D1. (a) LiF@Li10GeP2S12�Ƃ�ʾ��D��(b) TEM�D��(c) SAED�D��(d) HRTEM��EDS������D��(e) HAADF-STEM�D���Ԫ�طֲ��D��(f) F 1s XPS�D��
��D1a��ʾ����Li10GeP2S12��NH4F��ֻ������ǰ�(q��)�w������200 ���¼ӟᣬǰ�(q��)�w��(n��i)��NH4F�t���ܟ�ֽ�����NH3��HF��һ��Li10GeP2S12�w���cHF���w���|���͕��l(f��)�����·���(y��ng)��

ʹ���ܶȷ�����Փ��DFT��Ӌ��ó�ԓ����(y��ng)��500 K�µļ���˹�����ܞ�-1026.49 KJ mol-1��Li10GeP2S12�w���ı���������g�Ե�HFѸ�ٷ���������l(f��)�γ�LiF�⚤��ֵ��ע����ǣ���ԓ����(y��ng)�ж��Ե�NH3���w�������cLi10GeP2S12����(y��ng)������(y��ng)�a(ch��n)��GeF4��PF5���Ԛ��w����ʽ�]�l(f��)��
ͨ�^TEM�������D1b�����l(f��)�F(xi��n)LiF@Li10GeP2S12�w��������F(xi��n)��һ�Ӛ��ӡ�����̽��ԓ���ӵľ��w�ɷ֣����䚤���M��SEAD���D1c��������������7.02��4.97��4.30 nm̎�քe���F(xi��n)����������h(hu��n)�����Ԍ���(y��ng)��LiF�ģ�220������200���ͣ�111�����档
�Mһ�����w��߅���ֲ��Ŵ��M��HRTEM�������D1d����LiF@Li10GeP2S12�w��������F(xi��n)�˺�Ȟ�10nm�İ����ӣ����⣬EDS������D�D1d����HAADF-STEM�D�D1e���@ʾ��GeԪ�غ�FԪ�ص���������׃���������չ�F(xi��n)��ԓ�������ɺ�F(xi��n)Ԫ���γɵĻ�����D1f��Li10GeP2S12��LiF@Li10GeP2S12��XPSȫ�V�������@��LiF@Li10GeP2S12��684.9 eV��λ��̎���F(xi��n)��һ����LiF�����壬��C��LiF���ӵĴ��ڡ�
2. ��՚ⷀ(w��n)�����о�
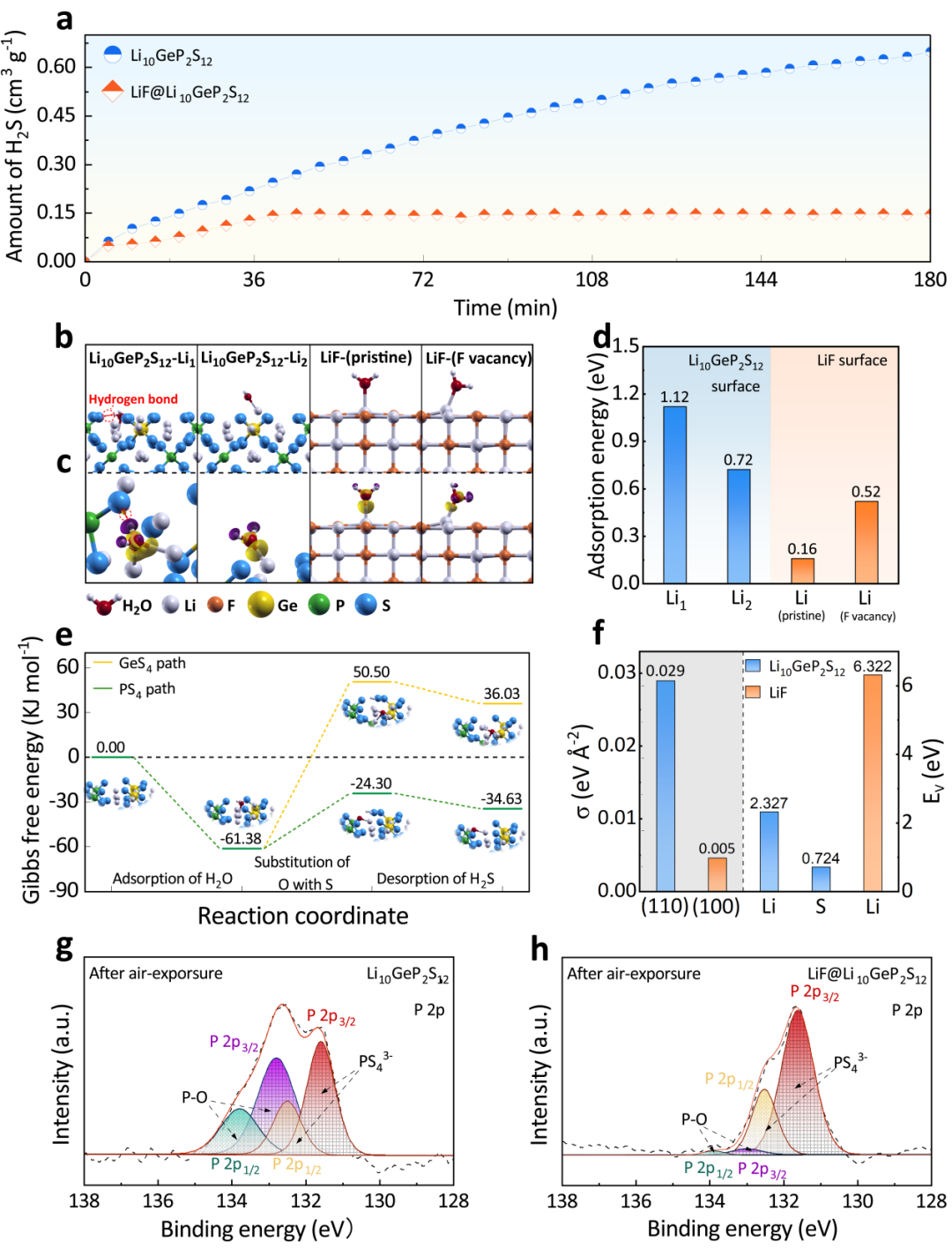
�D2. (a) Li10GeP2S12��LiF@Li10GeP2S12�ĕr�g-H2S���wጷ���������(b-c) Li10GeP2S12��LiF��������(g��u)�ͺ����P(gu��n)�������l(f��)�IJ��늺��ܶȣ�(d) H2O�ڲ�ͬ����λ�c�ϵ������ܣ�(e) H2O��Li10GeP2S12(110)�����ɷN����(y��ng)·������H2S�ļ���˹������׃��(f) Li10GeP2S12��LiF�ı����ܣ�σ���Ϳ�λ�γ��ܣ�Ev����(g) Li10GeP2S12�� (h) LiF@Li10GeP2S12��¶�՚�40min���P 2p XPS�����V��
��Li10GeP2S12��LiF@Li10GeP2S12��¶��30%������ȵĝ�՚��Ё��u����՚ⷀ(w��n)���ԡ���D2a��ʾ��LiF@Li10GeP2S12��H2S�������ʞ�0.00352 cm3 g-1 min-1����40����_�����ֵ0.1472 cm3 g-1����ֱ��180��状��]��H2S���w�^�m(x��)�a(ch��n)����H2Sጷſ�����Li10GeP2S12��4.4����
���������˽�Li10GeP2S12�ڝ�՚��е�ˮ��C����LiF�����ӵĝ�՚ⷀ(w��n)���ԣ�ϵ�y(t��ng)������DFTӋ����ԭ�ӳ߶���ˮ���ӵ������ͷ���(y��ng)�О顣���ȣ������ϵ�H2O������ˮ�ⷴ��(y��ng)�M�еĵ�һ���������Ժܴ�̶���Ӱ��˺��m(x��)PS43-ˮ�Ⲣጷ�H2S�ķ���(y��ng)��H2O���ӵ������ܣ�Eads�����x�飺
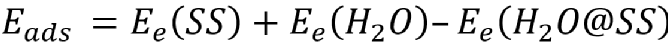
��������ʽ����������λ�cϵ�y(t��ng)�Y�x���l(f��)�F(xi��n)������Li1λ�c�ϵ�ˮ���ӿ����c����PS43-�γ�S-H�I���D2b-c��������@������ߵ������ܣ�1.12 eV�����S����S-H�I�ąf(xi��)���£�Oԭ�ӕ��cPS43-���F�е�Sԭ���ÓQ���������ʽጷŵ��՚��У��D2e����Ȼ������LiF�����ϣ�H2O�����ܵ���0.16 eV���y�Ԍ��F(xi��n)ˮ���ӵ�������
���⣬LiF��(y��u)�����淀(w��n)����Ҳ����ͨ�^���^�͵ı����ܣ�0.005 eV���C�����D2f������LiF@Li10GeP2S12�У�Li10GeP2S12���и�H2O�����ܵ�Liλ�c�ںܴ�̶��ϱ���ˮ��LiF�⚤����������Ч�p����H2O�����^�̺��S��PS43-��ˮ�⣬��(w��n)����LiF@Li10GeP2S12��(n��i)��Li10GeP2S12�ľ��w�Y(ji��)��(g��u)��
ͨ�^XPS̽����՚Ⱪ¶�������׃�����l(f��)�F(xi��n)Li10GeP2S12��132.9 eV��133.9 eV�Y(ji��)����̎���F(xi��n)�˃ɂ����壬��Դ��PS43-����ˮ��a(ch��n)����P-O�I���෴��P-O���P(gu��n)��ď�����LiF@Li10GeP2S12���V�D�ϘO��������LiF@Li10GeP2S12��PS43-��ˮ���^�١�
3. 늻��W(xu��)�����о�

�D3. (a) Li/Li10GeP2S12/Li��Li/LiF@Li10GeP2S12/Li���Q늳��R������ܶȜyԇ��(b) Li10GeP2S12��LiF@Li10GeP2S12�����늌�(d��o)�ʌ��ȣ�(c) Li10GeP2S12�� (d) LiF@Li10GeP2S12�ķֲ��B(t��i)�ܶȣ�(e) Li/Li10GeP2S12/Li��Li/LiF@Li10GeP2S12/Li���Q늳�ѭ�h(hu��n)���ܡ�
��D3a��ʾ���cLi/Li10GeP2S12/Li���Q늳���ȣ�Li/LiF@Li10GeP2S12/Li���Q늳ص��R������ܶȣ�CCD��ֵ�_����3.0 mA cm-2��ͨ�����f�����w늽��|(zh��)�^�͵����늌�(d��o)������Ч�Ĝp����x�������(n��i)��ֱ�ӳ��e�γ��֦�����M���@�ø��R������ܶȡ��Mһ��ͨ�^ֱ���O���yԇ̽�������늌�(d��o)�ʣ��D3b�����l(f��)�F(xi��n)LiF@Li10GeP2S12�����늌�(d��o)�ʞ�2.42×10-9 S cm-1�@������Li10GeP2S12�����늌�(d��o)�ʣ�1.09×10-8 S cm-1����
�ֲ��B(t��i)�ܶȣ�PDOS����ʾLiF�����ӵ�LiF@Li10GeP2S12���늌�(d��o)�ʽ��͵ă�(n��i)�ڙC������D3c-d��ʾ��һ���棬�K�w��LiF���б�Li10GeP2S12�����Ď�϶������LiF�Ľ^���Ը��ߣ���һ���棬Li10GeP2S12������б������^�ͣ�~21012 cm-2�����ԓ��s��Ѩ���@����һ������ӂ�ݔ��������LiF����Ď�϶��Ȼ�^����
���LiF�������܉���Ч���LiF@Li10GeP2S12����ӽ^���ԡ����⣬�^�͵����늌�(d��o)������Ч�p��늽��|(zh��)�c䇽��ٽ���̎������D(zhu��n)�ƣ��Ķ����ƽ��渱����(y��ng)�İl(f��)�������Li/LiF@Li10GeP2S12/Li ��0.1 mA cm-2/0.1 mAh cm-2���Ԍ��F(xi��n)1000С�r��(w��n)������x�ӳ��e/���x�����C�O��늉���±0.28 V��(n��i)��
4. ȫ�̑B(t��i)�늳ص�늻��W(xu��)�����о�
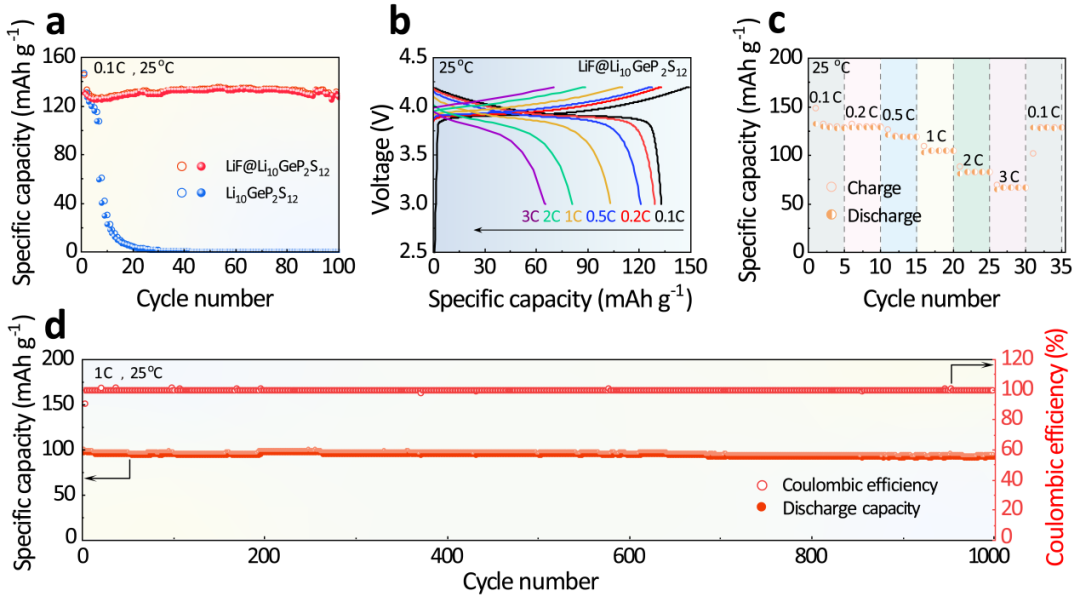
�D4. (a) LiNbO3@LiCoO2/Li10GeP2S12/Li��LiNbO3@LiCoO2/LiF@Li10GeP2S12/Li��0.1 C�µ�ѭ�h(hu��n)���ܣ�LiNbO3@LiCoO2/LiF@Li10GeP2S12/Li (b) ��0.1 C��3 C�µij��������Լ� (c) �������ܺ� (d) 1 C�µ�ѭ�h(hu��n)���ܡ�
�Mһ���u����LiF@Li10GeP2S12��ȫ�̑B(t��i)�늳��е�늻��W(xu��)���ܡ���D4a��ʾ��LiNbO3@LiCoO2/Li10GeP2S12/Li��7��ѭ�h(hu��n)������������ʃH��45.4%���y�Ԍ��F(xi��n)��(w��n)�����Lѭ�h(hu��n)����ʹ��LiF@Li10GeP2S12����늽��|(zh��)��ȫ�̑B(t��i)�늳ؿ��Ա��F(xi��n)��130.8 mAh g-1�ij�ʼ�������������100��ѭ�h(hu��n)��S��97.0%�ij������������ʡ�
�yԇLiNbO3@LiCoO2/LiF@Li10GeP2S12/Liȫ�̑B(t��i)�늳صı������ܣ��D4b-c�����l(f��)�F(xi��n)ȫ�̑B(t��i)�늳���0.1 C��0.2 C��0.5 C��1 C��2 C��3 C�·քe�����ṩ132.8��128.9��120.7��103.1��80.7��65.1 mAh g-1�Ŀ�����������
������LiF���ߵĽ����ܣ�LiF@Li10GeP2S12���Ԍ��F(xi��n)�����䇄��x/���e�����LiNbO3@LiCoO2/LiF@Li10GeP2S12/Liȫ�̑B(t��i)�늳ؿ�����3 C�ı����«@���^�ߵĿ����������yԇ1 C�����µ��Lѭ�h(hu��n)���D4d����LiNbO3@LiCoO2/LiF@Li10GeP2S12/Liȫ�̑B(t��i)�늳���1 C���܉���F(xi��n)��101.0 mAh g-1�ij�ʼ��늱���������1000��ѭ�h(hu��n)����F(xi��n)��94.8%�ă�(y��u)�����������ʡ�
�@Ȼ��LiF@Li10GeP2S12���ԘO��Ĝp���c�ؓ�O�g�ĸ�����(y��ng)������ڸ߱����«@���˃�(y��u)����ѭ�h(hu��n)��(w��n)���ԡ�
�C������������ͨ�^��������ķ����ϳ���һ�N����LiF�����ӵĺ˚����w늽��|(zh��)LiF@Li10GeP2S12��DFTӋ�������LiF�����Ӿ��ИO�͵�ˮ�����ܣ�0.16 eV��������Ч������Li10GeP2S12���λ�c�c�՚��е�H2O�l(f��)���������PS43-�ֽ⡣
���⣬LiF@Li10GeP2S12����2.42×10-9 S cm-1�ĵ����늌�(d��o)�ʣ�������Ч�����֦�����L������c�ؓ�O֮�g�ĸ�����(y��ng)�����ԓ늽��|(zh��)���R������ܶȏ�1.0 mA cm-2��ߵ�3.0 mA cm-2��
ʹ��LiF@Li10GeP2S12�M�b��LiNbO3@LiCoO2/LiF@Li10GeP2S12/Liȫ�̑B(t��i)�늳���1 C�¾���101 mAh g-1�ă�(y��u)�������������M��1000��ѭ�h(hu��n)����_94.8%�����������ʡ����������H��������w늽��|(zh��)��ˮ��C���ṩ���µ����⣬���Ҟ��O(sh��)Ӌ��՚��䇽��ٷ�(w��n)����������w늽��|(zh��)�ṩ���µ�ҕ�ǡ�
���˺���
ȫ�̑B(t��i)�늳�




��؟�������ăH�������߂����^�c���c�Ї�늳�(li��n)�˟o�P(gu��n)����ԭ��(chu��ng)���Լ�����������ֺ̓�(n��i)��δ��(j��ng)���W(w��ng)�C�����������Լ�����ȫ�����߲��փ�(n��i)�ݡ����ֵ��挍�ԡ������ԡ����r�Ա�վ�����κα��C����Z��Ո�x�߃H����������Ո���кˌ����P(gu��n)��(n��i)�ݡ�
�����W(w��ng)ע�� ����Դ��XXX�����Ї�늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո��һ�܃�(n��i)�M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@cbcu.com.cn
�����W(w��ng)ע�� ����Դ��XXX�����Ї�늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո��һ�܃�(n��i)�M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@cbcu.com.cn
����ϲ�g
-
�Ї��о��ˆT�_�l(f��)�����A(y��)䇻����g(sh��) ������x��늳ص�����
2022-12-29 08:32 -
���ݴ�W(xu��)�l(f��)���ºϳɷ��� ���������늳�늻��W(xu��)����
2022-12-15 08:42 -
Skoltech��������������O�Y(ji��)��(g��u)���·��� ��늳������ܶ�����25%
2022-11-14 09:13 -
��һ��ȫ���ׄ�(chu��ng)�ļ��g(sh��)�@��ͻ�ƣ�늳������ܶ�����3�������5���
2022-06-13 11:41 -
늄���܇�늳������ܶ�����10����ϣ�������Ӽ��g(sh��)�l�@��Ҫ�Mչ
2022-04-24 10:37 -
�v��(j��ng)10���о������늳ؼ��g(sh��)�K�ڳ��죬늄���܇���m(x��)������������
2022-04-06 10:40 -
�����ó�������܇늳�����ȡ�Ѓrֵ���� ������������97%
2021-10-15 09:34 -
�ձ�UNIST������x��늳��е���x������� ������늳ط������
2021-07-24 18:10 -
Sono Motors��Sino EV������늳ؼ��g(sh��)���m(x��)����̴������
2021-06-30 10:31 -
�о��ˆT�_�l(f��)����늽��|(zh��) ����䇿՚�늳�ѭ�h(hu��n)��(w��n)����
2021-05-14 08:50
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

���}


���P(gu��n)��
-
�Ї��о��ˆT�_�l(f��)�����A(y��)䇻����g(sh��) ������x��늳ص�����
2022-12-29 08:32 -
���ݴ�W(xu��)�l(f��)���ºϳɷ��� ���������늳�늻��W(xu��)����
2022-12-15 08:42 -
Skoltech��������������O�Y(ji��)��(g��u)���·��� ��늳������ܶ�����25%
2022-11-14 09:13 -
��һ��ȫ���ׄ�(chu��ng)�ļ��g(sh��)�@��ͻ�ƣ�늳������ܶ�����3�������5���
2022-06-13 11:41 -
늄���܇�늳������ܶ�����10����ϣ�������Ӽ��g(sh��)�l�@��Ҫ�Mչ
2022-04-24 10:37 -
�v��(j��ng)10���о������늳ؼ��g(sh��)�K�ڳ��죬늄���܇���m(x��)������������
2022-04-06 10:40 -
�����ó�������܇늳�����ȡ�Ѓrֵ���� ������������97%
2021-10-15 09:34 -
�ձ�UNIST������x��늳��е���x������� ������늳ط������
2021-07-24 18:10
�����c
-
�����_¶�ش���Ϣ��
2023-03-05 11:18 -
�[����ʮ�꣬�c�x��늳ؽK��Ҫ��̥�D(zhu��n)����
2023-03-16 09:32 -
������늳خa(ch��n)���^ʣ�������r������·�w��̎��
2023-03-07 08:58 -
����늳أ��l�ܷQ����
2023-03-26 13:02 -
����늳ؽ��r��(n��i)�����@�β�һ��
2023-03-14 11:07 -
��һվ������늳ػ���
2023-03-12 11:05 -
�Ј��O(ji��n)�ܿ��֣�����x��늳صȮa(ch��n)Ʒ��ʩ�����Ԯa(ch��n)Ʒ�J�C����
2023-03-21 09:27 -
��x��늳ظ������ܶ�ؓ�O�����о��Mչ
2023-03-12 11:06

©2017 ���(qu��n)���� 늳�(li��n)�� �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�rֵ�ɾ��ИI(y��)Ʒ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
�rֵ�ɾ��ИI(y��)Ʒ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I(y��)��̖
��I(y��)��̖ �Ź���̖
�Ź���̖