

�Ͽƴ������t&���������ӄ�AM: 500Wh/kg������Lѭ�h�����Եij��������ܶ�䇽���늳�

���������
�S������Դ�a�I�IJ�����£�����늳�ϵ�y�İlչ�������ϣ��������������ܶȵ�Ҫ��Ҳ�����������Ї��ƌW���g��(MOST)��������Դ��(DOE)���ձ�����Դ�a�I�C���_�l�C��(NEDO)���_����2030�����늳�ϵ�y�����ܶ����_��500 Wh/kg�@һ���_Ŀ�ˡ��@����ǰ��ʯī��ؓ�O����x��늳ض�����һ��������𣬶���ؓ�O��Q��䇽��ٺ������ܶ���Ѹ�����40%���ϣ��@ʹ��䇽���늳ء����늳ؼ�䇿�늳،��ɞ����L�ڄ���늳�ϵ�y�����е������wϵ��
�M��䇽���늳������O����^����Փ�����ܶȣ�����Ҏģ���Ås���R��������K������Ҫ��ԭ�����ڸ��Ե�䇽���ؓ�O�ڳ����^���Е��c늽�Һ�l�����������²��ɿص��֦�����L����䇣�������䇻�����IJ������ɡ��֦�����^���γɕ��Ɖ�ؓ�O���w늽��|����Ĥ(SEI)���̴���Ĥ�������صğ�ʧ���M�����l늳�ȼ����ը����Щ���Q�֦������Ҫ���Դ��w�ɷ֞����N��(1) ʹ�øߝ����}늽�Һ�p����x�ӵIJ�����ֲ���(2) ����늽�Һ���ӄ�����SEI��(3) ���ؓ�O���昋���˹�SEI��(4) �OӋ3D�{�Y����䇽���ؓ�O��(5) ��늽�Һ��Q��̑B늽��|��(6) �γ�䇺Ͻ�LiX(X=C��Si��)�p���֦�����L���M���@Щ���Զ�ȡ����һ���ij�Ч�����oһ���Ԡ��������ܶ�������r��������ȷ���µ�ѭ�h�������x����Ŀ����������h��
���ɹ����顿
���գ��Ͽƴ������t&���������ӄ���Advanced Materials�ϰl�����}��“500 Wh kg−1Class Li Metal Battery Enabled by a Self-Organized Core–Shell Composite Anode”�����¡�ԓ����ͨ�^��Һ�B����LM(GaInSn)��Ϳ�ھ۱�ϩ(PP)��Ĥ���M��䇳��e���l�γ���Ӟ�䇻���Һ�B����(LixLMy)���ȌӞ�䇽��ٵĺ˚�ؓ�O�Y����ԓ�Y���������õ��x�ӌ�ͨ�ʺ�늌��ʣ�����NCM622���O��ȫ늳�չʾ��1500Wh L-1��483Wh kg-1�������ܶȣ�����NCM811���O���Mһ���������ܶ�������514 Wh kg-1.
������c��
1. ��Һ�B����LM(GaInSn)��䇻��^�����l�γɵ�䇻�Һ�B����(LixLMy)����䇽���ؓ�O����ȱ��o�ӣ����Է�ֹ䇽����c늽�Һֱ�ӽ��|���c����������ȣ�LixLMy�������õ���x�ӌ�ͨ�Լ���늌��ʡ�
2. ԓ䇺˚��Y��ؓ�O����NCM622���Oչʾ��1500Wh L-1��483Wh kg-1�������ܶȡ���Һ�B���ٺϽ�����γɵ�SEI���ИO�ߵķ����ԣ�ÿȦѭ�h�����������ʸ��_99.95%��
3. Li������LixLMy�ֲ��������L���B�m��䇽��ٱ��ӣ���Ч�ر������֦���Įa�������C��늳ص�ѭ�h�����Լ���ȫ�ԡ�
���D���x��
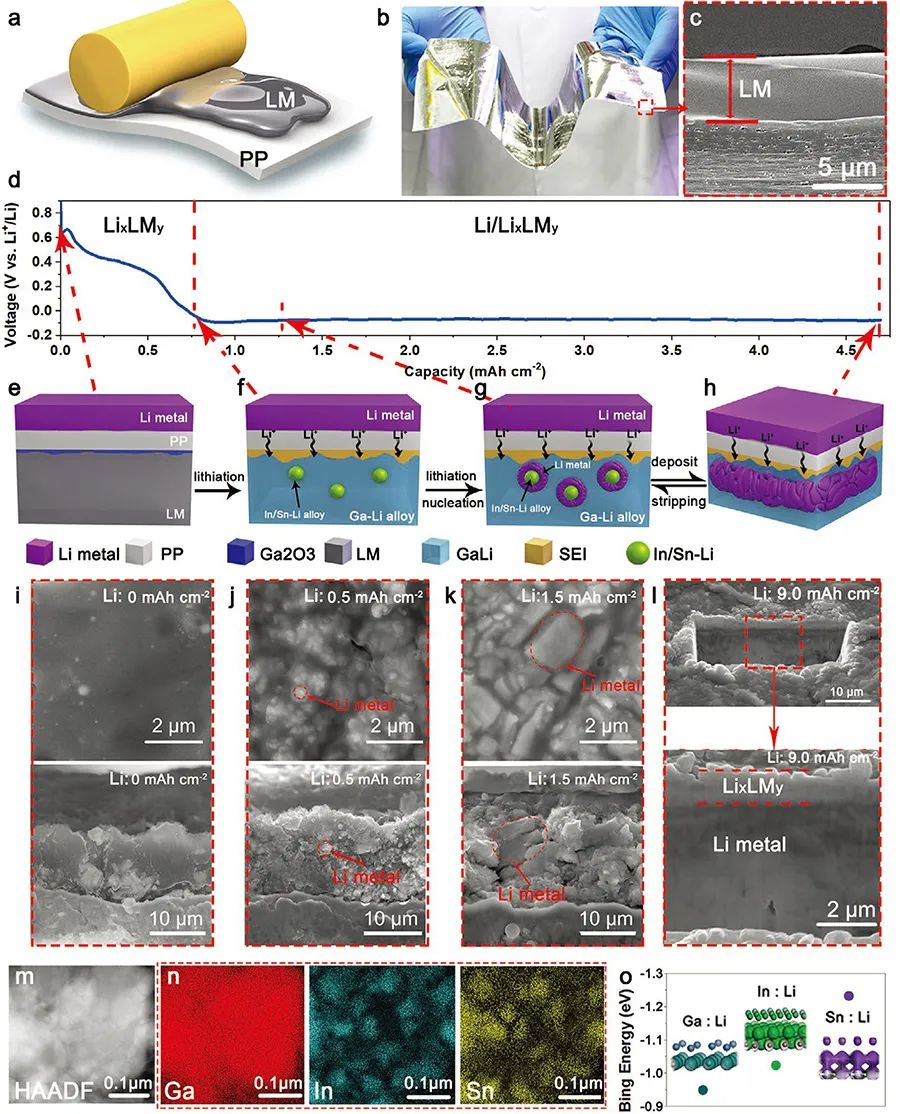
�D1. �ͺ�늘O(Li/LixLMy)���γ��^��
(a) ͨ�^��Ϳ�γ�LM��Ĥ��ʾ��D��
(b, c) LM��Ĥ�Ĺ�W��Ƭ������SEM�D��
(e-h) �ͺ�늘O���γɈD�⣻
��i-l) ��ͬLi������Li/LixLMy��SEM�ҕ�D�ͽ���D��
(m) LixLMy������STEM�D��
(n) m�D��ͬһ�^���Ԫ�ط���, Ga(�tɫ), In(�{ɫ), Sn(�Sɫ)��
(o) Li�cGa, In, Sn�ĽY����ģ�M�Y����
��D1(a)��ʾ���ͺ�늘O��ͨ�^Һ�B����LM��䇻��^��һ���ϳɡ����ȣ�5���LM(GaInSn)�Թ�Ϳ�ķ�ʽ�����ھ۱�ϩ(PP)��Ĥ�ϡ�LM������������ʹ�������PP���M�о����Ҿo���İ�������D1(b,c)������LM/PP�cLi���M�b��늳غ�ͨ�^䇻����ÿ����l�γ�Li/LixLMy�ͺ�늘O���ϣ��D1d�����ɺ��������������Կ������ڷ���������_0.75 mAh cm-2֮ǰ��늄�Ѹ���½���֮��څ�ڷ����M��ƽ�_�ڡ���D1(e)��ʾ������Ga��LM�е�Ѹ����������PP�cLM�Ľ���̎�����Fһ��Ga�ı������ӡ�LM�c Li�ĺϽ���������LixLMy�����ɣ��l����䇻��^�̵������A�Σ��D1(f,i)������늄���1.0 VѸ�ٽ���0.1 V�@һ�^�̡���Ȥ���ǣ����γɵ�LixLMy�Юa�����l�ķ����^�̡���STEM��Ԫ�ط����ɿ������ڸ�Ga���|�����������^�쵽Ƕ�����еĸ�Sn��In�u��Y�����D1(m,n)���@�ɚw����Li�cSn��In�ĽY�������^��Ga���Ը��ߣ��D1(o)������ˣ�Li���A�����cSn/In�γ�SnInLi�F�أ��M�����F���ࡣ���@֮���M��LM�����xLi�x�ӄt���l��SnInLi�F���ϳɺ����L���D1(g,f)��������u�γɱ�GaLi���������B�m䇽��ٱ��ӣ��D1(h,l)����
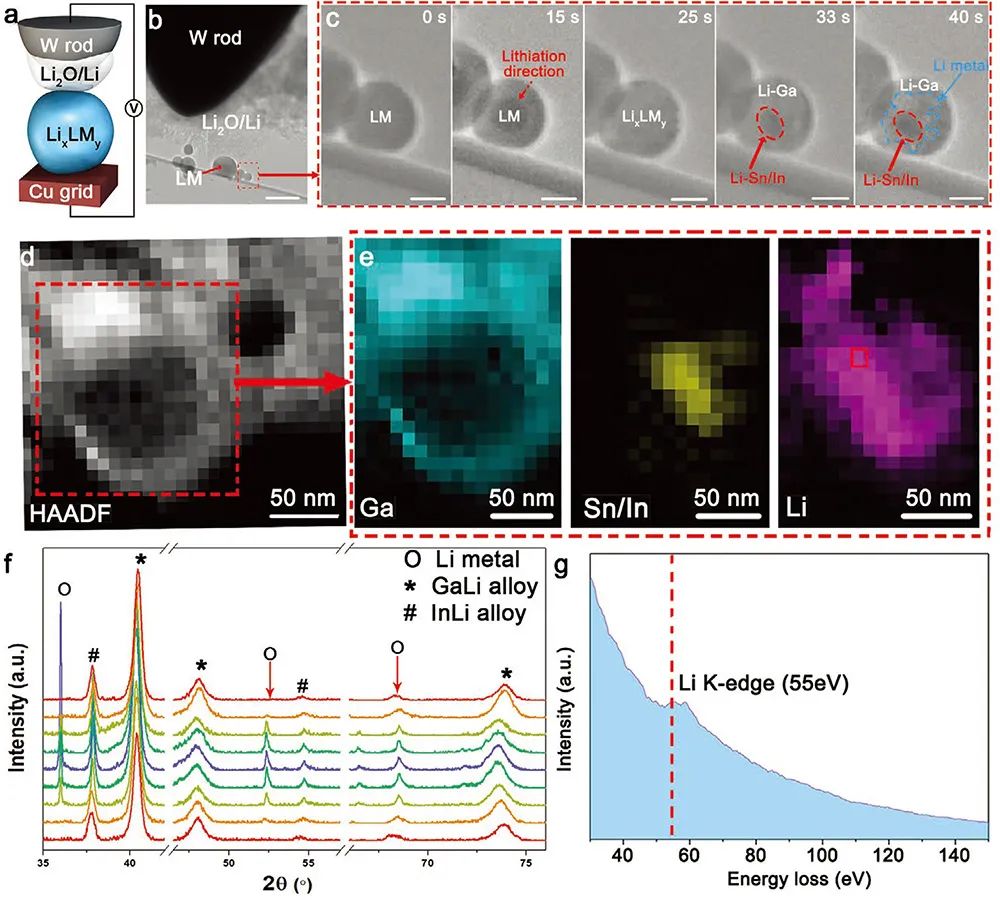
�D2. ԭλTEM��XRD�о�Li/LixLMy���γ��^��
(a) ԭλTEMʾ��D������̼֧��Ĥ�ϵ�LM�{���w���cᘠ��u늘O��Li/Li2O늽��|���־o�ܽ��|��
(b) �c�D(a)��������TEM�D��
(c) �S��Li���e�r�g���ӣ�LM�{���w����ԭλ����TEM�D��
(d) LM�{���w����STEM�D��
(e) ��������pʧ�V(EELS)Ԫ�ط�������Li/LixLMy�ɸ�Li����ɫ����Sn���Sɫ���ăȺ˼���Ga���{ɫ�����⚤�M�ɣ�
(f) ̎�ڲ�ͬLi���e�A�ε�Li/LixLMyԭλXRD�о���Li����������0������9 mAh cm-2, �S����0��
(g) ��EELS�õ���Li K-edge�V����Ϣ��
����ͨ�^ԭλTEM��XRD��Li/LixLMy�ͺϽY�����l�γ��M���������о���STM��Ʒ�U�ɰ���Li��ᘠ��u늘O������̼֧��Ĥ�ϵ�Һ�B����LM�M�ɣ���D2(a,b)��ʾ����Li������Ȼ�γɵ������ӿ�����̑B늽��|���c�������ɈD2(c)�ɰl�F�����^15s��Li���e��LM���ⲿ�^���_ʼ׃�ù�����25s���_��Ƕ�͡��S��Li���|��GaLi���|���_ʼ�ɺˣ�33s�������L��40s��������γ�Li/LixLMy���߽ǭh�ΰ�����HAADF������������pʧ�V(EELS)�����Y���Mһ���C����Li/LixLMy��Ȟ鸻Ga�^�����g�鸻Sn����In�^��Li���e�^����D2(d, e, g)�������⣬����߀�l�F�S��Li�ij��e��Ó����Li/LixLMy�ͺϽY�����γ���һ�������^�̡���D2(f)�е�ԭλXRD��ʾ���ڰl��Ƕ�֮ǰ��XRD�V�Dֻ���^�y��GaLi��InLi���V�壬�S��Li�IJ���Ƕ����e��Li������ď����S֮��������LiÓ�������V����u�p���������^��GaLi���V����]�аl��׃��������GaLi���Ӿ������õķ����ԡ�
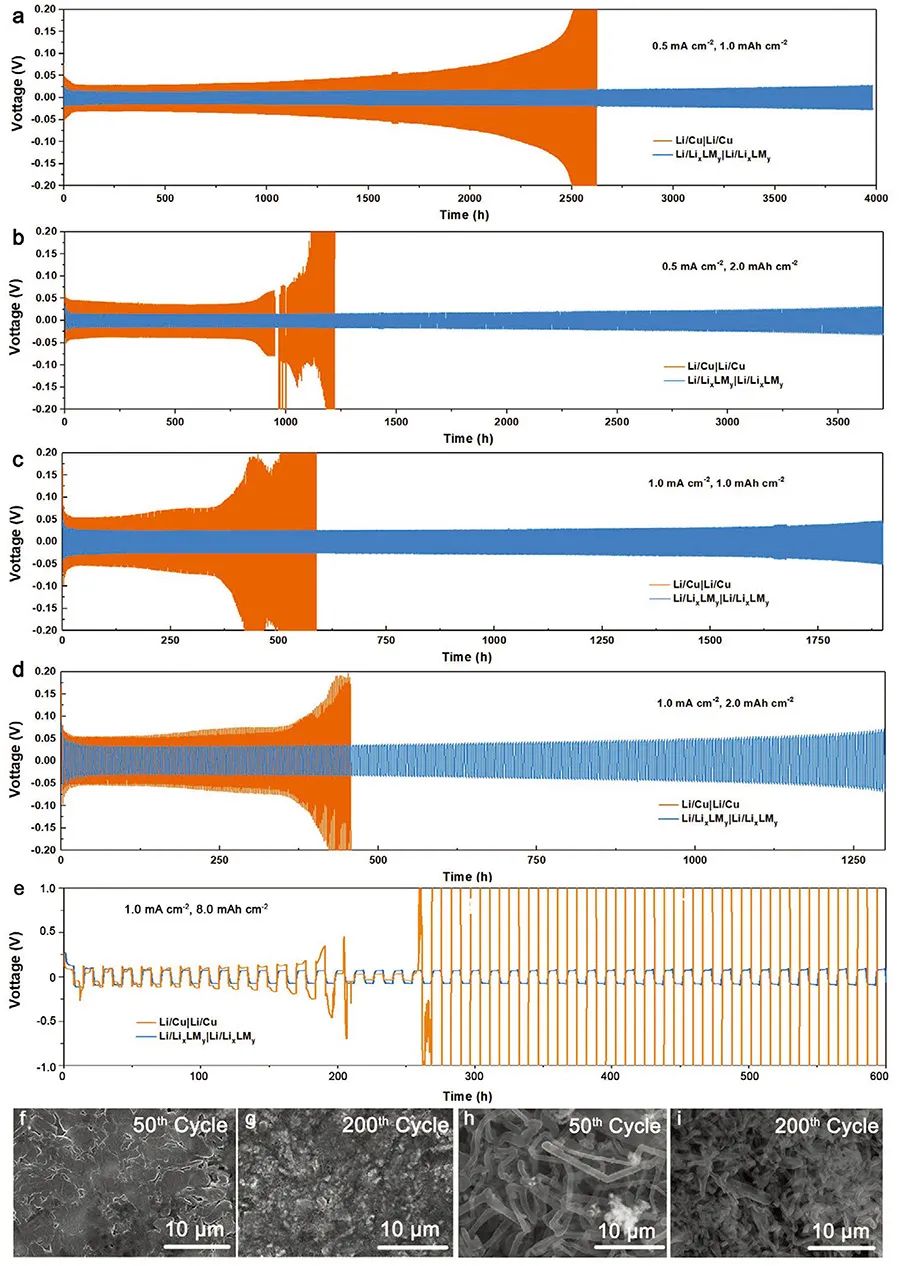
�D3. Li/LixLMy�Lѭ�h��늻��W����
(a-e) ��ͬ����ܶȼ�������Li/LixLMy�cLi/Cu��늘O�^늄��ȣ�늽�Һ��1 M LiPF6 in EC:EMC���w�e��1��1�����~������1 wt% VC����
(f, g) Li/LixLMyѭ�h50Ȧ��200Ȧ���SEM�D��
(h, i) Li/Cuѭ�h50Ȧ��200Ȧ���SEM�D��
Li/LixLMy�ĸ߿����Ա��C��ԓ�ͺϽY���ڂ��y늽�Һ�l���µ��Lѭ�h�����ԡ������Ԍ��Q늘O(Li/LixLMy|Li/LixLMy)�����о�������һ�M��һ��늘O��Q��Li/Cu���酢�ȣ���0.5��1.0 mA cm-2������ܶ����о����ߵ�늻��W�О顣��D3��ʾ�����нY�����C���� Li/LixLMy���и��͵��^늄ݼ���������ѭ�h�����ԡ����^3700С�r�ij���ѭ�h��Li/LixLMy���^늄ݵ���30 mV���D3b������늘O����]���֦���Įa�����ֲڶȱ����ژO�͵�ˮƽ���D3f, g����������ͬ������ܶ��£�Li/Cu늘O���^늄ݳ��^Li/LixLMy�ɱ����ұ��沼�M�֦��������ѭ�h1200С�r��ʧЧ���cǰһ�M�γ��r�����ȡ�
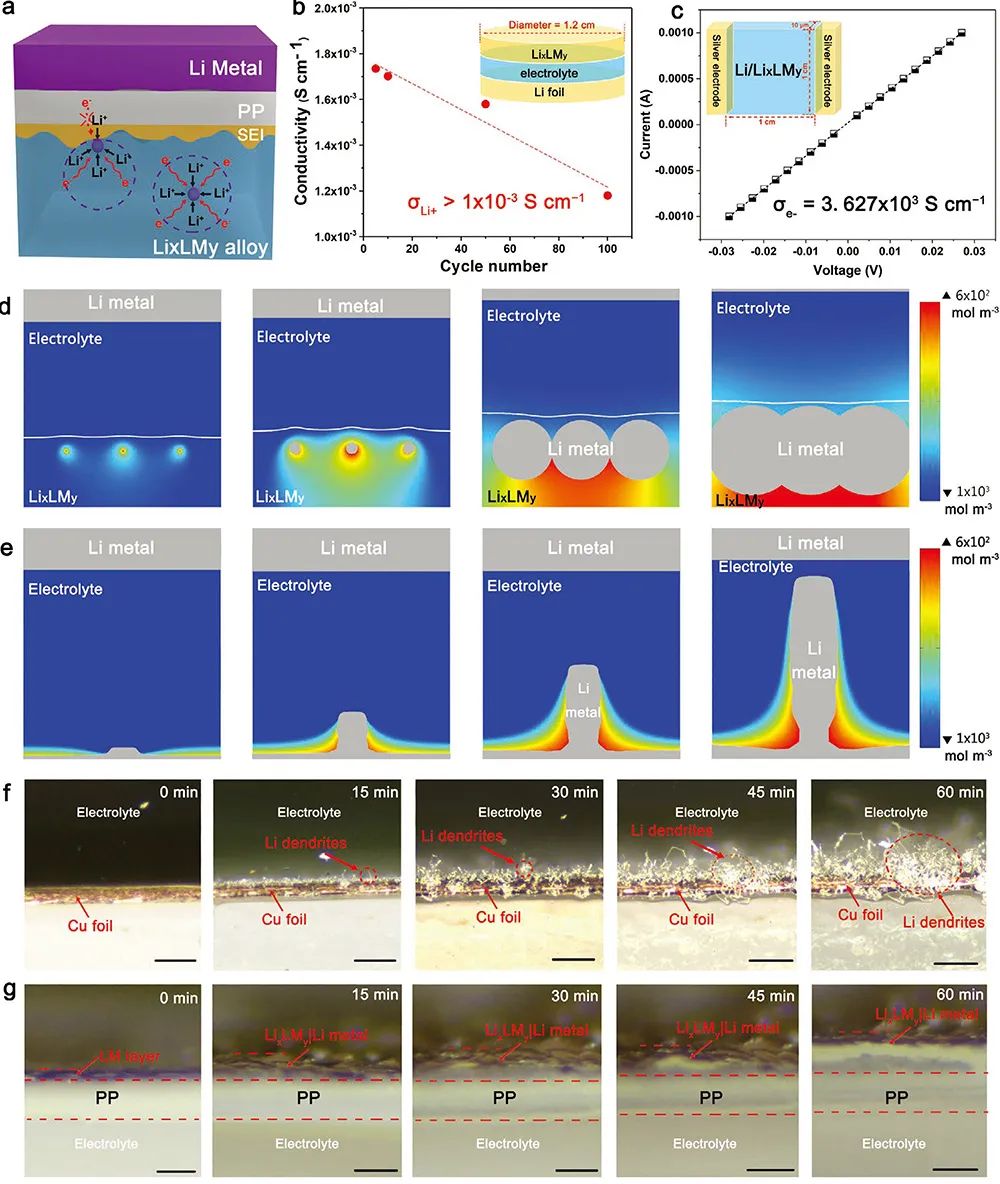
�D4. Li/LixLMy�o֦�������о�
(a) LixLMy��Li���ٸ���ͬ�����Lʾ��D��
(b, c) LixLMy���x�ӌ�ͨ�ʡ�늌��ʜyԇ��
(d, e) LixLMy��Cu��Li���ٳ��e�^�̵��ΑB��׃ģ�M��
(f, g) 2.0 mA cm-2������ܶ��£���Cu����LM��ͨ�^늻��W���eLi�Ĺ�W�@�R�D�����ߞ�20�ס�
Li/LixLMy늘O������Ч�������֦�������L���@��Ҫ��������Ȍ�늱��o�ӵ������������á����⣬�Ȳ�GaLi���|���x�Ӻ�����ṩ�����õČ�ͨ�h������ʹ��x�ӿ����ڏͺϽY���о�����e���Ķ��Mһ�������γ��֦������D4(a)�����ɈD4(b)�ɿ�����ѭ�h��GaLi���ӵ��x�ӌ�ͨ���Կ��_��1.0*10-3 S cm-1��ͨ�^���±����y�õ�GaLi늌���Ҳ���_3.63*103 S cm-1���C����GaLi���п��ٵ���x�Ӻ���ӂ�ݔ�����W���ԡ��D4(d, e)չʾ������COMSOLģ�M��Li��Li/LixLMy���~���ϵij��e�О顣��Li/LixLMy�ͺϲ����У�С�w��Li���˾���ֲ�����u���L����w���M���γ��B�m��Li���ӡ�����Cu���ϵij��e�О�t�c֮�෴������Li���˳ɺ�λ�c������С�w��LiѸ�ٳɞ�����ľ�횣��Ķ��a���֦����ģ�M�Y���cԭλ��W�@�R�D���Ǻϣ��D4f, g������2.0 mA cm-2������ܶ��£�60��犺�Cu���ϱ鲼횠���֦������LM����t���^�y��������e��Li���ӣ��������͵�ÓǶ��^�����m���տs����Û���f��LixLMy����ͬ�r��䷀���Լ����g�ԡ�
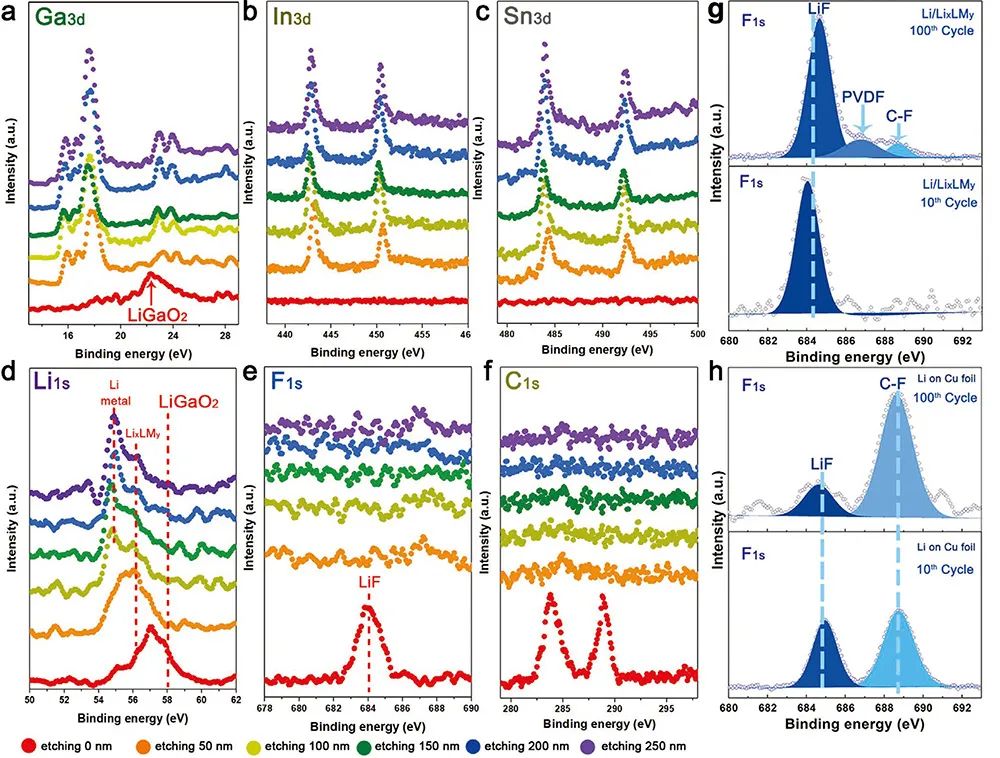
�D5. LixLMy��SEI�ķ������о�
(a-f) ��ͬ���g���XPS�D�V(a: Ga3d; b: In3d; c: Sn3d; d: Li1s; e: F1s; f: C1s)��
(g) 10Ȧѭ�h��100ѭ�h��SEI�ӵĸ߷ֱ�XPS�D�V��
(h) ���Ռ����Li/Cuؓ�O��SEI�ӵĸ߷ֱ�XPS�D�V��
�ɈD5(a, f)�ɿ�������늠�B�ͺϲ��Ϗ��ϵ��µĽM�ַքe��SEI�ӣ�LixLMy��LixLMy�е�䇽��١�δ���gǰ��F1s�D�V���D5(e���tɫ�������f����늘O��늽�Һ�Ľ���鸻LiF��SEI�^��LiF���γ���Ҫ�w����늽�Һ����x�ӵ�߀ԭ��ͬ�r������Li�x���cGa2O3�ķ���ʹ��SEI����߀����LiGaO2���D5(a, d�����������g��Ȟ�50 nm�r��F 1s�еļ����ʧ��ȡ����֮����Ga 3d��In 3d��Sn 3d�@��LixLMy�����壬�f��LiF�cLiGaO2�H������LixLMy��픲����S�����g��ȵ����ӣ�Li���|�ĺ���Ҳ�S֮���ӡ��D5(g, h)�ĸ߷ֱ�F1s�D�V�f��SEI�ό���LiF�ĺ��������ЙC��}������100Ȧ��׃���������f����LixLMy���隤�Ӻ��SEI��ѭ�h�^����ʮ�ַ������෴����Li/Cuؓ�O�У�����Liֱ�ӽ��|늽�Һ����늽�Һ����߀ԭ��100Ȧ��LiF�ķ及�@�������ЙC��}����ζ�����yLi����ؓ�O�ϵ�SEI����ѭ�h�^���ИO��������
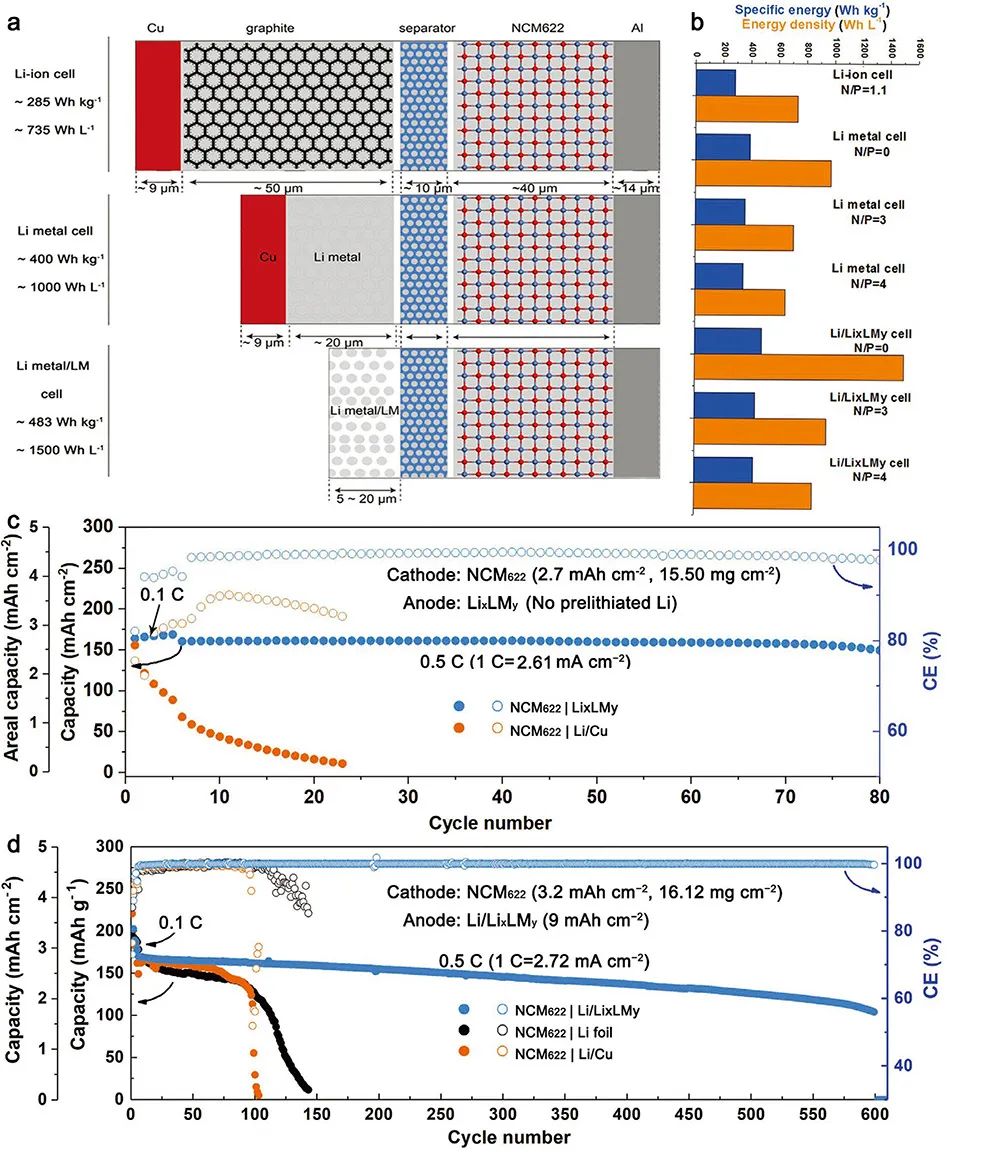
�D6. Li/LixLMyؓ�O�ij��������ܶ��о�
(a) NCM622������yʯīؓ�O���ϲ�����䇽���ؓ�O���в�����Li/LixLMyؓ�O���²�����ԭ��ʾ��D��
(b) ��ͬN/P����NCM622|ʯī��NCM622|䇽��٣�NCM622|Li/LixLMy늳ص������ܶȣ�
(c) ��N/P�Ȟ�0�rNCM622ȫ늳ص�ѭ�h����;
(d) ��N/P�Ȟ�2.8�rNCM622ȫ늳ص�ѭ�h���ܡ�
����LixLMy/PP�ĸ�늌��ʼ���ƥ�����ԣ�NCM622|LixLMyȫ늳��в�����Ҫ�~������ؓ�O�����w���@������Ч�p��늳ص��������w�e���Ķ��Mһ������ȫ늳������ܶȣ��D6(a,b)����
��N/P�Ȟ�0�r����ζ��LixLMy�]���M���A䇻�����NCM622|Li/LixLMy늳ص������ܶȿɸ��_1500Wh L-1��483Wh kg-1���ǂ��yNCM622|ʯī늳ص�2.04����ͬ�r���ڳ��^80Ȧ��ѭ�h�����^���У�ÿȦ�����������ʿɸ��_99.12%���D6(c)������N/P�Ȟ�2.8�r����0.5C��1C=2.72 mA cm-2��������ܶ��£�500Ȧѭ�h�����������ʞ�75%, �����ܶȞ�437Wh kg-1������yNCM622늳ص�1.5�����D6(d)����
�����Yչ����
䇽���늳���������֦�������_���y�Ա��C늳صİ�ȫ�ԏĶ��ڌ��H���������y���أ�������Һ�B����LM����|���w��ͨ�^䇻������l�γ�LixLMy���ӣ��M���ڃȂȾ������L��䇽��ٱ��ӣ���K�γɺ˚��ͺϽY������늌��ʡ��߷����Բ�������g�Ե�LixLMy������Ч�ظ��^��䇽����c늽�Һ��ֱ�ӽ��|��ͬ�r�o���~�����鼯���w����Ч�pС��늳ص��������w�e������Li������LixLMy�о���ֲ������ԣ�ԓؓ�O��ѭ�h�^���п����M�о����䇳��e���Ķ������֦���Įa�������c����NCM622ƥ��r�������ܶȿɸ��_1500Wh L-1��483Wh kg-1������������ѭ�h�����ԡ�������NCM811�������O���ϕr�������ܶȿɳ��^500Wh kg-1���M��ͨ�^䇻������Ƃ��Һ�B���ٺϽ�ؓ�O�^�y��Ҏģ���a�����@һ�OӋ��Ȼ��������ܶ�䇽���늳��ṩ�ˌ��F��˼·��
���īI��Ϣ��
500 Wh kg−1 Class Li Metal Battery Enabled by a Self-Organized Core–Shell Composite Anode (Advanced Materials, 2020, DOI: 10.1002/adma.202004793)
�īI朽ӣ�https://onlinelibrary.wiley.com/doi/full/10.1002/adma.202004793
(؟�ξ�������)




�����Wע�� ����Դ��XXX�����Ї�늳��ˣ�������Ʒ�����D�d������ý�w���D�dĿ�����ڂ��f������Ϣ�������������Wٝͬ���^�c�͌����挍��ؓ؟��
������Ʒ���ݡ������������}��Ҫͬ���Wϵ�ģ�Ո��һ�܃��M�У��Ա��҂����r̎����
QQ��503204601
�]�䣺cbcu@cbcu.com.cn
-
�Ϻ��ƴ�AM��ʯ��ʯ��늽��|����ȫ�̑B䇽���늳�
2020-12-10 10:19 -
�Ї��ƴ��늳ع̑B늽��|�C���о�ȡ����Ҫ�Mչ
2020-04-17 08:37 -
�Ї��ƴ��@�������늳ط�����
2020-04-16 10:15 -
�Ї��ƴ�����������늳ؿ��_������
2019-12-16 10:57 -
�Ї��ƴ�¹������༉�Y�����Ƹ����ܹ̑B�늳؏ͺ�ؓ�O
2019-06-10 09:52 -
�ձ�᪸�ˎ�ƴ��аl���ܷ�ֹ�����ӻ��ĸ�Ч�ƚ似�g
2019-02-20 11:05 -
�Ї��ƴ����Ƴ������|�ӽ��QĤȼ��늳�ꎘO����
2018-12-05 09:03 -
���ƴ��������B�ЙC̫���늳��о��I��ȡ���Mչ
2018-03-06 13:41 -
�Ї��ƴ�����ׂ����m���_�P���ЙC����̫���늳��OӋ
2018-03-02 10:06 -
�Ї��ƴ�̼������늴������о��@�Mչ
2018-01-23 14:14
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



-
�Ϻ��ƴ�AM��ʯ��ʯ��늽��|����ȫ�̑B䇽���늳�
2020-12-10 10:19 -
�Ї��ƴ��늳ع̑B늽��|�C���о�ȡ����Ҫ�Mչ
2020-04-17 08:37 -
�Ї��ƴ��@�������늳ط�����
2020-04-16 10:15 -
�Ї��ƴ�����������늳ؿ��_������
2019-12-16 10:57 -
�Ї��ƴ�¹������༉�Y�����Ƹ����ܹ̑B�늳؏ͺ�ؓ�O
2019-06-10 09:52 -
�ձ�᪸�ˎ�ƴ��аl���ܷ�ֹ�����ӻ��ĸ�Ч�ƚ似�g
2019-02-20 11:05 -
�Ї��ƴ����Ƴ������|�ӽ��QĤȼ��늳�ꎘO����
2018-12-05 09:03 -
���ƴ��������B�ЙC̫���늳��о��I��ȡ���Mչ
2018-03-06 13:41
-
2020���늳��ИI�о����
2021-05-11 11:24 -
ͻ�l������һ����Iֹͣ���I����ɢ�T����
2021-05-11 10:02 -
4���҇�����늳��b܇��ͬ������134.0%
2021-05-13 08:26 -
��ο���Pack���F䇺���Ԫ����
2021-06-01 09:25 -
�ɳ�������\�՚�늳أ��Mչ�������δ��
2021-05-19 10:59 -
���_�M�h䇘I�ļҵ�
2021-06-03 09:46 -
��늳ػġ����u�����a��늄���܇��܇��Ҫ���������ˣ�
2021-06-01 21:22 -
�P����������x��늳����B�mʽ��ո���ϵ�y���gҎ�����ȃ���ИI�˜ʵĺ���������Ҋ����
2021-05-31 22:53

�rֵ�ɾ��ИIƷ�ƣ����\�����ṩ���������YӍ
��ICP��09081210̖
 ��I��̖
��I��̖ �Ź���̖
�Ź���̖